The
Development and Design of a Generic Genome
Analysis
Environment: The G-language system
Arakawa Kazuharu
Faculty of Environmental Information,
Abstract
The short but grand history of
bioinformatics has clarified the fact that it must gain higher efficiency in
order for it to process the massive information that it faces in the future.
This paper introduces an
attempt to solve the task which bioinformatics faces today by:
1.
Constructing
an analyzing software developing environment aiming for the integration of the
various individually developed conditions.
2.
Systematically
accumulating existing analysis software techniques for analysis and their
results.
3.
Constructing
generic analysis packages which allow users to avoid redundancy in the process
of analysis.
This document discusses the
philosophy and the significance of the G-language project, and states in
detail, the development and design of its G-language System.
1. Introduction
1.1 The
significance of the G-language Project
The
reading of the draft sequence of the human genome has finally been completed
last year, bringing us to the opening of the post genome era. This signifies that the importance of
genome analysis, which searches for an ultimate understanding of life through
analyzing digital data consisted of the four letters “a t c g”, will continue
to increase.
Bioinformatics has been taking a great
roll in science since the complete genome sequence of Mycoplasma Genetalium was
made public in 1996, but its short history has clarified the fact that its
systems must gain higher efficiency in order to process the massive information
that it faces in the future. Making
new discoveries by exploring the massive genome data which is now constantly
being produced requires a great amount of time and human resources. In order to reconstruct the
procedures of bioinformatics to have higher efficiency, the following points
must be attained.
1. The construction of an environment for
the development of integrated analysis software. Such software is now usually being
developed individually under various conditions, with no common specifications.
1.
The
Systematic accumulation of existing analysis software, procedures and their
results.
2.
The
construction of generic packages of analysis procedures that allow users to
avoid redundancy in the process of analysis.
The first method saves
time and cost necessary for the development of the new software. Bioinformaticians who develop their own
applied software for analyzing genome information usually need to develop their
own versions of software for the most basic, commonly used methods. Not only is this way generically
inefficient, but it is also undesirable in terms of the flexibility of the
software produced, in addition to its deficiency in ability for it to be shared
and recycled. This causes an
unnecessary burden on developers, and wastes a great part of the resources in
the field of bioinformatics. A
generic development platform suited for genome analysis which supports basic
methods is capable of dramatically improving the efficiency of software
development.
The second method allows researchers to
choose and combine methods and software which match individual purposes by
accumulating a pool of tools in a public domain within an integrated
development system. This way of
increasing efficiency in research is only possible because bioinformatics uses
various recyclable methods that are stored as software, a form fit for easy
copying and distributing.
Connecting the pool of tools with bioinformatics databases, which are
constantly enriched on the internet, will make it possible to make use of
existing research results to the greatest extent.
The third method increases efficiency by
constructing generic packages of analysis procedures which are otherwise
repeated by handwork. Non-generic
software designed to comprehensively analyze basic data of the genome of a
specific specie is likely to be useless for other species. If such a package were to be developed
on the generic integrated platform, countermeasures for unexpected difficulties
can be taken easily simply by reconstructing part of the comprehensive analysis
package. The integration of
existing analytic methods also have a great potential to be widely applied to
new researches.
The attempt to construct a
bioinformatics system actively rational with the conscious of the three points
raised above is unprecedented. This
document discusses the G-language Project, which attempts to construct
such a system, and Prelude (The developing code of The G-language
System’s core version 0.1 to 1.0).
1.2
Background
Before explaining the details
of the G-language Project,
projects similar
to it are summarized below in order to clarify the significance and the tasks
that The G-language Project faces.
Bioperl
The
Bioperl project attempts to make an integrated development environment as a
group of Bio:: modules written in the programming language “Perl.” It pays close attention to its functions
such as its methods of input/output, coping with various formats of array
databases, provides a number of useful methods along with genome information
structs, and even using virtual memory when handling huge amounts of data like
the human genome. It also includes
modules that use routine tools such as BLAST and FASTA for analysis. The Bioperl project fulfills the first
point mentioned in the previous section of this document, but it does not work
for many irregular types of the Genbank database format, nor dose it have a
reliable accuracy in parsing genome annotation data. It lacks functions to fulfill the goals
of the G-language Systems.
For example, it has little conscious to provide bioinformatics analysis
tools other than those used most commonly.
Biopython, Biojava, Biocobra and Bioruby are projects similar to
Bioperl.
EMBOSS
EMBOSS is a set of genome
analysis software provided by the Norwegian EMBnet. EMBOSS methods and software for analysis
are substantial in both number and quality, and has a simple standard for
coding new software, enabling it to be used on various command lines. This project too, though, has defects
from the standpoint of the G-language Systems goals. It has no concept of an integrated
development environment, nor does it have a basic platform.
Darwin
In
order to overcome the tasks that the projects stated above have yet settled,
the G-language Project began in the year 2001, at the Institute for
Advanced Biosciences of Keio University.
At this point, development of prototype version 0.5 has been
completed. The release of version
1.0-beta is planed in October on the internet http://www.g-language.org/.
2
Program architecture
2.1 Design concept
When
designing the G-language system, a generic genome analysis environment, the
flexibility of the system should be most taken into consideration. If many and
unspecified genome analysis methods and contents are developed by the unified
system on the assumption that they are reused, a more flexible system design is
desired. Moreover, it is indispensable that the various genome database files
and the existing open source software can be broadly used in this system.
The program
architecture of G-language System was designed as shown in Fig. 1, in which the
flexibility of the system was taken into consideration as the first priority.
This design consists of a three-layer structure, i.e. a user interface layer, a
script module layer, and a core layer. Each layer is built as a completely
separable independent system. Since various input and output make a structure
object which consists of unified formats in a core layer, and the structure
object itself is delivered to a script module layer, the data through the core
layer can be treated without taking in account its form in a script module
layer. In addition, by unifying the output from a script module layer, the
result analyzed by various programs can be viewed without troubling with the
different forms of the user interface layer.
The meaning
of making G-language System into the independent three layer structure is not
only the advantage of the communication between programs described above but
also the simplicity of exchange of a layer in the system according to the
situation. Bioperl, which is capturing the spotlight in the similar projects as
described in the preceding chapter, has a function equivalent to the core of
G-language System. Making each layer become independent in a design stage makes
it possible to perform analysis by the script module layer using the core of
Bioperl in G-language System. This enables a user to take in outstanding
functions from the exterior flexibly and to make the favorite combination of
systems. The epoch-making points of G-language System as an integrated system
of bioinfomatics are that the various analysis techniques are accumulated in
its script module layer, and that it has flexibility very much.
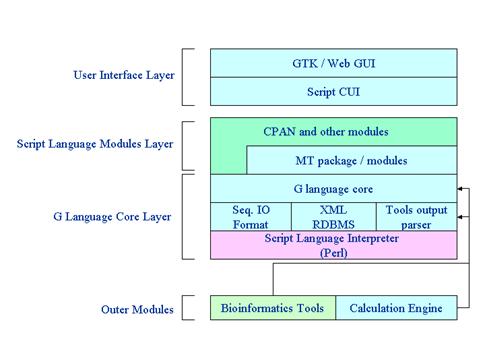
Fig. 1 Program architecture of
G-language System
2.2 Architecture
The substance
of G-language System is the module group of script language Perl [8]. Perl was
developed by Larry Wall [9] and has been most generally used as a script
language. Perl has a very flexible description and many useful modules accumulated
by the Internet archive groups, such as CPAN [10]. In order to give flexibility
widely as development environment, it becomes a big advantage that programming
is easy and flexible and that there are many existing modules. Moreover, a
script language which is good at character sequence processing is suitable for
analyzing the genome database which is a document so to speak [11]. Thus the
usage of a script language using rather than a compile language is advantageous.
However, it may
be desirable for the core to be built by a compile language from a viewpoint of
calculation efficiency, or by script languages which are more developed, such
as Pyton or Ruby. Although these points had essential aspects, the G-language
Project, in consideration of the situation such that the present developer is
presently unfamiliar in languages other than Perl, the development language was
unified to Perl.
The most
general genome database format is a Genbank-form flat file database format
[12]. Input and output of the other formats can be processed by supporting
readseq, a database format conversion tool, in the core. Moreover, from a
viewpoint of the amount of data and reference speed, the following strategies
are adopted in the G-language System: a relational database can be used as
standard, which is accessed by Pg modules in CPAN and gbk2pg, a part of Prelude
core. The G-language System core holds data which pass these file input and
output inside a program as a structure object, and it is used in a script
module layer.
3 Mounting of G-language System
3.1 Prelude
Among the G-language
System core layers, Prelude consists of (1) the G class which holds database
information as a structure object as described in the preceding chapter, (2)
the gbk2pg class interlocked with a relational database, and (3) the Rcmd class
which communicate with R language, a statistics analysis language. The G class
is a super class of all the systems of G-language System, and MT package in a
script module layer is loaded as a library and a module in this class. That is, the instance of the G class
inherit G class automatically and can use the function of the MT package as a
native function. Thus, the false program language that analysis is performed,
which extends the system using a Perl language in the development environment
of G-language System, is built by offering a diversified function from MT
package. In the future, this false program language environment will be
extended to a more advanced one by the script type user interface.
The
flexibility of G-language System makes it possible to replace a core layer by
Bioperl as described in the preceding chapter. However, there is a big meaning
in daring develop Prelude, considering problems such that Bioperl is not
designed as an integrated system and does not support R language which is
useful in statistics analysis. In
addition, its structure object with database information cannot respond to the
irregular form of Genbank. Now, Prelude can conquer these problems and play
role sufficient as a core of a bioinfomatics integrated system, which posseses
many fundamental techniques of genome sequence analysis such as intron, exon,
and so on as a native function are supported as a native function, and holds a
memory which can be operated with the pointer of a file handle so that a huge
database format such as human genome can be treated. The Perldoc documentation explaining
the detailed function of G class is appended to the end of the book.

Fig. 2 Prelude core in
G-language System
3.2 MT package
The MT
package is a set of the various bioinfomatics analysis techniques, and is
constituted as several modules which are collected based on functional relation
in the inside. As described in the preceding chapter, a user is provided with the
MT package as a native function because it is loaded as a library from G class,
a super class from the inherited class and an instance of Prelude.
Furthermore,
the relation between the functions in MT package is the homage. That is, the function, in which the
several functions of the minimum unit (for example, which calculates GC skew)
are created and packed, and performs batch processing. The bacterial complete
coverage analysis system, which is mounted in the present G-language System,
integrates/unifies the function in this MT package and is itself offered as a
function of MT package. The integrated system software exists based on the
design of such a general-purpose script module layer.

Fig. 3 Outline on MT
package
3.3 Example of System
Mounting: MT::System::Mutation_finder
An example of
system mounting is shown below in order to introduce how the G-language System
functions. The flow of “MT::System::mutation_finder”
method, a system which explores mutation information from two different
bacterial genome databases, is shown in Fig. 4. (1) sequence homology analysis by BLAST
is performed for every gene from the genome data, (2) mutations of homologous
genes are investigated based on the result, and (3) the site of the mutation and
its influence on translation and secondary structure are analyzed and
outputted.
The
mutation_finder method of the MT::System class is called on the G-language
System, and the genome data structure object is read during Prelude activation.
Based on these data, the mutation_finder method calls another MT method groups
inside, performs each analysis, and returns an output. Furthermore, each method
such as MT::uni-all_blaster, MT::bi-bi_clustalwer, and MT::RNAfolder calls
BLAST, ClustalW, and RNAfold which are the external programs from the inside,
respectively. If complicated processing is realized under the unified
environment where the MT package is the general-purpose method group based on a
Prelude structure object which
constitutes a multiplex structure, development cost will decrease and
the reusability of the individual method will increase. This hints a
possibility that the MT method located in the lowest layer is used as
composition parts of a different system.
Because input and output of the MT::System
are the Prelude structure object which is common to the G-language System, it
can be performed without troubling to the file of a different format, and is
effective as a general-purpose analysis system.

Fig.
4 Program figure of
MT::System::mutation_finder
4.
Discussions
4.1 Plans for further
Development
The development of The G-language
System is now at version 0.5; There are still many tasks to be tackled
before the beta release in October:
l
The
preparation of documentation
l
The
development of user friendly UI
l
Exhaustive
function checking
l
The
construction of analytic packages from existing MT modules
l
The
enrichment of the MT::System class
There must be an additional increase of
basic analysis tools prior to the The G-language Systems formal release
in December.
4.2 Distinction
At the opening of the post genome era,
many projects similar to the G-language
Project are launched. Therefore
the G-language Project must become distinctive from the others. Providing a system replete with
high quality applications, solving the 3 tasks which bioinformatics faces stated
in chapter 1, focusing on constructing a system to satisfy both low end and
high end users (not intermediate users), and inviting developers from the
public, will be the most important keys to the success of the G-language
Project.
In order to clarify the merits
of this unprecedented generic analysis environment constructed by combining two
independent pieces of software, Prelude and the MT modules, it is necessary to
arrange the UI environment to make the system open to low end users.
The G-language System is available
to the public domain (GPL) with an open source copy left concept, and the G-language
Project invites you to be part of the development of its system.